Self Crosslinking Polymeric Dispersants Used in Emulsion Polymerization
Several properties are important for high-performance coatings, including the following.
- Hardness and scratch resistance
Anti blocking (for stacking recently coated substrates)
Resistance against household chemicals and grease
Outdoor durability and UV resistance
Flexibility and toughness

Surfactants in Emulsion Polymerization
Surfactants play a crucial role in emulsion polymerization. They are required for emulsification of the monomers, formation of micelles as polymerization loci and colloidal stabilization of the polymer particles. In addition, surfactants reduce the surface tension of the resulting dispersion, which is required for wetting of a substrate and for film formation. With surfactants, dispersions can be formulated into a coating because the surfactants will help avoid destabilization of the dispersion with the addition of formulation components. In fact, they may even be an aid to effective mixing of these components and the dispersion.Surfactants are, in most cases, water soluble and mobile components in the film. They have a tendency to cluster together or migrate (see Figure 1), either to the film-air interface or the film-substrate interface.1 In Figure 2, surfactant exudation to the surface of an acrylic dispersion film is shown, where the exuded surfactant crystallized at the polymer air interface. The surfactants can in this way seriously affect the water sensitivity of the film, as well as the adhesion characteristics.
Surfactants that remain on the interfaces of the polymer particles will create hydrophilic channels through the film, which may cause undesirable effects such as water transport through the film to the substrate.
Several options have been studied to reduce the negative effects of surfactants while maintaining the positive aspects, such as the use of copolymerizable surfactants1-2 and polymeric surfactants. Copolymerizable surfactants will be chemically bound to the polymer particles and cannot migrate through the film or to the surface freely. As a result, films from acrylic dispersions prepared with copolymerizable surfactants should have better water resistance and adhesion properties.
There are, however, some issues associated with the use of copolymerizable surfactants. The reactive groups of these surfactants
are often not of the same reactivity as the reactive groups of the monomers used, which may lead to poor incorporation or
homopolymerization of the surfactants in the water phase. On the other hand, if incorporation is good the resulting dispersion can
have high surface tension because there is no free surfactant left, which impairs wetting, leveling and formulation of the
dispersion. To promote good copolymerization with the acrylic monomers, the reactive group of the surfactant should be in the
hydrophobe of the surfactant, and such surfactants are hardly commercially available.

It is also possible to prepare surfactant-free acrylic dispersions, which are solely stabilized by the anionic initiator (persulphate) end groups,4 but in general only low-solids systems can be prepared this way. The solids can be raised by adding ionic monomers, but generally the resulting systems have low tolerance toward the addition of formulating component.
Another route to obtaining surfactant-free dispersions is the preparation of self-dispersing polymers in organic solvents, which are subsequently dispersed into water.5 After dispersing, the organic solvents are distilled off to yield a secondary dispersion or artificial latex. Here the dispersing groups are usually acid groups that have been neutralized to salts by the addition of an amine. The ionized polymers have molecular weights of 10K-50K, are surface active and are known to be useful in the stabilization of hydrophobic polymers. In this case, core-shell particle morphologies are created where the ionized polymer forms the shell, and the hydrophobic polymer forms the core of the particle. The ionized polymer in this case functions as the stabilizer for the hydrophobic core.
Upon film formation of such secondary dispersions, the matrix of the film will consist of the ionized polymers and
hydrophobic particles are dispersed in this matrix. Since the ionized polymers are usually low in molecular weight and contain
fairly high levels of copolymerized and ionized acid, the matrix of the film will tend to be the weakest part of the system with
regard to sensitivity to water and toughness of the film. The low molecular weight of the ionized polymers will aid the coalescence
process and entanglement of the polymer chains to build up strength in the film.

Film Formation
Proper film formation6 is crucial to achieve a good performance of the final film. The film formation process can be divided in several stages7-8 (see Figure 3) where the entanglement step especially will determine the final properties. During the entanglement process, the interface between initially separate particles will gradually fade away. The strength of the film, both in mechanical and in resistance terms, will increase during the entanglement process.Acrylic dispersions generally have very high molecular weights, much higher than conventional solventborne polymers, such as alkyds. This is beneficial for the overall film properties, but makes the entanglement step very slow. The buildup of properties in acrylic dispersion films may, therefore, take days or even longer. In some cases, optimal film formation is never achieved because the coalescents that keep the polymers mobile evaporate before entanglement is complete.
Especially high-Tg and high-molecular-weight acrylic dispersions can achieve very good application properties, but for good film formation, high levels of coalescents are required. With the ongoing drive toward more environmentally friendly coatings, the aim is to reduce this coalescent demand further with zero VOCs as the ultimate goal. In recent years, several options have been investigated and published aimed at combining the good properties of high-Tg polymer backbones with good film formation.
One method is the use of a very hydrophilic acid and, in some cases, low-molecular-weight polymer backbone. If a
sufficient amount of acrylic or methacrylic acid is incorporated into the backbone, these dispersions can be either alkaline soluble
or alkaline swellable.9-10 In both cases, the minimum film formation temperature (MFT) and the coalescent demand are
significantly reduced. To maintain a workable balance between viscosity and solids content, low-molecular-weight polymers or
oligomers are used. Since low-molecular-weight polymers are not very water- and chemical-resistant, and hydrophilic backbone
polymers have poor water resistance, they are often combined with some form of crosslinking after or during film formation.
Clearly the poor resistance properties are the most important drawback of this approach.
Applying Crosslinking to Improve Chemical Resistance
While good MFT-hardness balance can be achieved with hydrophilic oligomers, this approach has the disadvantage of inferior chemical resistance. This can be overcome by post crosslinking the low-molecular-weight polymer. Crosslinking can be effective in two ways: chemical reaction between polymer chains following interdiffusion and crosslinking at the interfaces of the particles. In the first case, a coherently formed film is fixed and the molecular weight of the chains present in this film is increased. In the second case, two particles are chemically bound by crosslinking during or just after film formation. Formation of a chemical link between particles has a similar effect on properties, such as entanglement between polymer chains, but can take place on a much shorter time scale. A variety of crosslinking reactions have been described in past years. There are so-called two-component crosslinking systems where one of the crosslinking components is added just before application of the dispersion. More desirable for this work are self-crosslinking (one-pot) systems where all reactive components are present and long-term storage stable. The crosslinking reaction can be triggered by the evaporation of water upon drying, a change of pH, or by curing at elevated temperature, where the crosslinking reaction is faster, or reactive groups are de-blocked. Examples of suitable crosslinking systems are the reaction of amines with epoxy functionality11 where either can be on the polymer backbone, the auto-oxidation of incorporated fatty acid groups,12-13 the self condensation of alkoxy-silane functionality,14 the self condensation of n-methylolacrylamide,15-16 metal-ion co-ordination with backbone functional groups such as acetoacetoxy15 groups or acid groups, and the reaction of acetoacetoxy groups with amines17-18 or acetoacetoxy groups with unsaturated groups in a Michael reaction.19Results and Discussion
This article discusses the use of polymeric self-crosslinking dispersants for the preparation of surfactant-free acrylic dispersions20 with a core shell morphology.The core consists of normal high-molecular-weight hydrophobic acrylic polymer. One purpose of the shell is to stabilize
the core against flocculation and coagulation. To do this, the shell is made up from high-Tg hydrophilic alkaline-soluble polymer
chains that are modified with hydrophobic components to make them highly surface active (such polymers will be referred to as
polymeric dispersants).

Polymeric Self-Crosslinking Surfactant
Figure 4 compares the surface tension of aqueous solutions of such polymeric dispersants to polymethacrylic acid and a commercially available hydrophilic alkaline soluble acrylic oligomer and a commercially available styrene-acrylic acid oligomer. As can be observed, at very low concentration the surface tension of the polymeric dispersant solution is quickly reduced and equilibrates. The final surface tension is approximately 43 mN/m, while for polymethacrylic acid this is >60 mN/m.This is likely caused by the multiple hydrophobic groups that a polymeric dispersant contains, which causes the polymer chain to stretch along the surface of the air water interface. The figure shows that using a commercially available hydrophilic alkaline polymer is far less effective in reducing the surface tension.
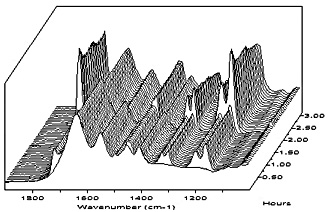
To demonstrate that the self-crosslinking polymeric dispersants are effective stabilizers for the preparation of acrylic
dispersions, a REACT-IR experiment was performed. The REACT-IR probe detects small particle size (monomer swollen)
particles and water-dissolved monomers. Large monomer droplets are outside the detection range of the REACT-IR probe.
Monomer is added to a solution of self-crosslinking polymeric dispersant in which the probe is immersed.



For comparison, a similar REACT-IR experiment was done with sodium lauryl sulphate as the surfactant (see Figure 6), and also with a commercially available styrene-acrylic acid oligomer as the stabilizer (see Figure 7).
Figure 6 does not show a monomer peak, nor does any reaction take place after addition of the initiator. This can be
explained by the stirring of the experiment, which is done by a simple flat-blade stirrer at low rpm. While this method of stirring
was adequate for emulsification of all monomer when the self-crosslinking polymeric dispersant was used, it proves insufficient
for proper emulsification of the monomer by SLS. Visually the reaction mixture does not become white or milky, but stays a bit
gray with clear inhomogenity. With this kind of inhomogenity it is clear that no small monomer droplets will be created.



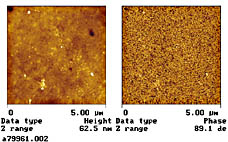
To verify that a core shell structure is obtained, the dispersion was analyzed with TEM. The self-crosslinking polymeric
dispersant was modified so it could be selectively stained vs. the core polymer. Figure 8 shows that the stained self-crosslinking
polymeric dispersant (the dark colored material) is present in the particle shell and in the water phase.

The Effect on Film Formation
This hard hydrophilic self-crosslinking polymeric dispersant will have good film formation since it is an alkaline-soluble polymer. Entanglement, however, is not applicable in the self-crosslinking polymeric dispersants, since their molecular weights are just below the entanglement Mw.Consequently, a very fast mixing of the low-Mw hard hydrophilic polymeric dispersant will occur. This is then followed by a crosslinking reaction leading to a crosslinked continuous hard phase containing discontinues soft domains. The formation of crosslinks at the particle interface will give an even better buildup of film strength as polymer chain entanglement would, and generally at a much shorter time scale. By focusing crosslinking at the particle interface it is used most effectively. Many of the crosslinking techniques are expensive, and should be used efficiently. Precrosslinking should be avoided as much as possible, since this will negatively affect entanglement during film formation.
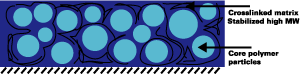
Alkaline-soluble systems on their own generally lack sufficient resistance properties to be useful. To overcome this
problem, we have used the polymeric dispersants as stabilizers in a subsequent emulsion polymerization. This way, core shell
particles are created that have the polymeric dispersant as the shell, and a high-molecular-weight resistant polymer in the core (see
Figure 9).
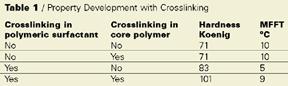
As can be observed, crosslinking of the core polymer alone has no effect on the hardness MFT balance, compared to no
crosslinking at all. If just the polymeric dispersant is self-crosslinking the hardness MFT balance is improved. But the best result
is achieved if both the polymer and the polymeric dispersant crosslink.

Film Formation and Properties of Model Dispersions
In the system under investigation there are several design parameters that can be adjusted to optimize application properties.- Ratio between shell material and core material
- Tg of both phases
- Hydrophilicity of the core
- Acid value of the shell phase


Entry 7 yielded a very good balance between MFT and surface hardness, 15 deg C and 121, respectively. Reducing the acid value of the shell led to a marginal decrease in surface hardness while the MFT increased with 20 deg C.
These results show that shell:core ratio, Tg of the high-molecular-weight core, and acid value of the shell can be useful tools to control the balance between film formation on the one hand and film properties on the other.
For most applications, anti-blocking is a required property. Normally, an easy way of introducing anti-blocking is to
increase the Tg of the film, which will cause poor film formation properties. For our model emulsions with a shell:core ratio of
1:1 good film formation could be combined with excellent blocking properties. In Table 4 blocking properties of model emulsions
are shown as a function of chemical composition of the core. For both core compositions very good blocking and early blocking
results were obtained. Interestingly, these good blocking properties were obtained for films containing 50% by weight of very
low Tg material. Table 4 shows that at these low core Tgs good surface hardness properties were found and acceptable MFT's.
Especially the polymer having a core containing methyl methacrylate gave a very interesting set of properties, with an MFT of
only 15 deg C, high surface hardness and excellent blocking properties.

Application in High-Performance Coatings
Variation of the design parameters can have a big impact on the application properties. When the design is optimized for application in industrial wood coatings the following properties can be achieved (see Table 5). Because of the absence of surfactant the system will be low foaming. The dispersion has a very low particle size, which results in excellent film formation and high transparency. An additional benefit of the low particle size is the almost translucent optical appearance of the binder, which is comparable to traditional solventborne coatings. Also wood wetting, which is an important property for industrial wood coatings, is excellent. This is caused by the good wetting potential of the self-crosslinking polymeric dispersant and the small particle size of the latex, which makes penetration into wood pores efficient.By modifying some of the design parameters, a surfactant-free self-crosslinking acrylic dispersion for joinery can be
created. In this application, flexibility is of importance combined with good anti-blocking properties (see Table 6). For achieving
good outdoor durability the coating will have to be flexible enough to compensate for the changing wood dimensions, which are
weather dependent. The coatings need to be fast drying and should be applicable by a variety of application techniques, such as
spraying and dipping. Again these dispersions have a very low particle size and the desired translucent appearance.

The dispersions based on self-crosslinking polymeric dispersants can also readily be used as binders for high-performance inks. The alkaline-soluble polymeric dispersant provides good reversibility to the system, a key property for use in inks. When the ink is dried further and crosslinking will take place the final ink will be highly resistant even to ammonia, where for conventional inks this is commonly an issue. The core shell morphology provides the combination of good block resistance combined with very high flexibility, which makes these systems also very useful for application on flexible substrates such as PE films. An added advantage is that these dispersions have excellent adhesion properties to treated PE film, even after prolonged water exposure. The self-crosslinking polymeric dispersant provides very good wetting properties to various substrates, and makes high pigment loadings possible to give high-strength inks. High-solids systems (55-60% solids) can be prepared with this technology that have the benefit of very fast drying. Table 7 shows some of the properties that can be achieved in an ink binder based on this technology.
While in this article the focus has been on high-Tg shells and low-Tg cores, the use of low-Tg shells combined with high
Tg cores can also lead to very interesting systems for a variety of end uses.
Conclusion
Self-crosslinking polymeric dispersants are efficient stabilizers for use in emulsions polymerization, giving good wetting properties and paint formulation stability to the resulting polymer dispersion. Contrary to conventional surfactants, they do not foam or migrate through the final film - two very beneficial features. When high-Tg self-crosslinking polymeric dispersants are used for the preparation of a core/shell polymer where the shell contains the self-crosslinking polymeric dispersant stabilizing a hydrophobic high-Mw core polymer, a very good MFT/hardness balance can be achieved. Here the self-crosslinking polymeric dispersant brings high performance to the coating, forming the crosslinked matrix where a conventional surfactant would only degrade the properties of the coating. Because of their superb film forming properties combined with crosslinking features the resulting core/shell dispersions require little or no coalescent, and yet give excellent film formation, hardness, anti-blocking and resistances against chemicals, making such dispersion very useful for high performance coating and ink applications.21Acknowledgement
The authors would like to thank Danny Visser, Harriet van der Sande, Dorina van Haeringen, Anton Peters Marc Roelands and Guru Satguru for their contributions to this work.This article won an Outstanding Paper Award at the 2002 International Waterborne, High Solids, and Powder Coatings Symposium in New Orleans. Symposium sponsored by The University of Southern Mississippi Department of Polymer Science.
For more information on dispersants, contact NeoResins, Sluisweg 12, PO Box 123 5140AC, Waalwijk, The Netherlands; or
visit www.NeoResins.com.
References
1 A Schultz et al. The use of non-migrating surfactants in emulsion polymerization; Proc. of the 3rd biennial North American res. conf. on the science and techn. Of emulsion pol / pol coll. (1999).2 Holmberg, K. Prog. Org. Coat., 20 (1992) 325-337.
3 Bouvy, A. Eur. Coat. J. 11/96 822-826.
4 Juang, M.S.; Krieger, I.M.; J. Pol. Sci.: Pol. Chem. Ed. Vol. 14, 2089-2107 (1976).
5 Schlarb, B.; Gyopar, M.; Rau, S. Haremza; Prog. Org. Coat., 26 (1995) 207-215.
6 Peters, A.; Overbeek, G.; Buckmann, A.; Padget, J.; Annable, T. Prog. Org. Coat. 29 (1996) 183-194.
7 Winnik, M.; Wang, Y.; Haley, F. J. Coat. Techn., Vol. 64, No. 811, August 1992.
8 Daniels, E.S.; Klein, A.; Prog. Org. Coat., 19 (1991) 359-378.
9 Jin, Z.Z.; Zhu, Y. J. Coat. Techn., vol. 60, No. 757, February 1988.
10 Padget, J.C. J. Coat. Techn., Vol. 66, No. 839, December-1994.
11 Geurts, J.M.; v Es., J.J.G.S; German, A.L. Prog. Org. Coat. 29 (1996) 107-115.
12 Chen, F.B.; Bufkin, B.G. J. Appl Pol. Sc.,Vol. 30,4551-4570 (1985).
13 Pulin, P.; Raval, D.A.; Mannari, V.M. J. Coat. Techn. Vol. 70, No. 883, August 1998.
14 Bourne, T.R.; Bufkin, B.G.; Wildman, G.C.; Grawe, J.R. J. Coat. Techn. Vol. 54, No. 684, January 1982.
15 Bufkin, B.G.; Grawe, J.R. J. Coat. Techn., Vol. 50, No. 641, June 1978.
16 Bufkin, B.G.; Grawe, J.R. J. Coat. Techn., Vol 50, No. 643, August 1978.
17 J. Feng et al. J. Coat. Techn. Vol. 70, No. 881, June 1998.
18 Esser, R.J.; Devona, J.E.; Setzke, D.E.; Wagemans, L. Prog. Org. Coat. 36 (1999) 45-52.
19 Clemens, R.J.; Del Rector, F. J. Coat. Techn. Vol. 61, No. 770, March 1989.
20 Buckmann, F.; Overbeek, A.; Nabuurs, T. Eur. Coat. J. (2001), (6), 53-60.
21 This technology is protected by several patents, owned by NeoResins.
Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!



