UV-Curable, Hybrid Organic-Inorganic Coatings
Dr. Darlene Brezinski presented the Outstanding Coatings Technologies First Place Paper Award, sponsored by PCI, for $3,000 to Bret Chisholm at the 30th Annual International Waterborne, High-Solids, & Powder Coatings Symposium[R] in New Orleans.
For many applications, thermoplastics possess many desirable properties such as low weight, high impact strength and good processability. However, a major drawback of thermoplastics over metals, ceramics, or thermosets is low scratch and mar resistance. As a result, many thermoplastic applications require a hard coating to impart abrasion resistance to the article. As a means to increase the hardness and abrasion resistance of coatings for plastic substrates, the addition of organically modified silica particles to organic coatings has been investigated.1
The first organically modified silicates were independently prepared by Philipp and Schmidt2 in 1984 and Wilkes, Orler, and Huang in 1985.3 These new inorganic-organic hybrids were named “ORMOSILs” by Schmidt and “Ceramers” by Wilkes. The term ORMOSIL has since been used extensively to describe organically modified silicate nanoparticles.4-5
A variety of organic groups have been grafted onto the surface of silica particles using organosilanes. For example, Chu et al.6 prepared epoxy functional silica particles by condensing glycidoxypropyltrimethoxysilane on to the surface of colloidal silica particles. These ORMOSILs were used to produce coatings by curing with a polyamine-curing agent. Hunnicutt and Harris7 studied competitive surface reactions between binary organosilane mixtures and silica particles to determine differences in silane reactivity as a function of alkyl chain length for a series of haloalkysilanes. Becker et al.8 prepared thermoplastic nanocomposites based on ORMOSILs produced from the modification of silica particles with methacryloxypropyltrimethoxysilane and acetoxypropyltrimethoxysilane. The effect of silica surface modification on the thermomechanical properties of the composites was investigated. The objective of the work described in this article was to investigate the affects of ORMOSIL composition and matrix composition on the properties of UV-curable, organic-inorganic hybrid clear coatings on polycarbonate. The organosilanes chosen for the investigation provided insight into the affect of grafting the ORMOSIL nanoparticles to the organic matrix. In addition, the matrix materials were chosen to illustrate the impact of the ORMOSIL nanoparticles on matrices ranging from lightly crosslinked, rubbery materials to highly crosslinked, glassy materials.
Experimental Materials
The colloidal silica used for the study was Snowtex ST-O-30 from Nissan Chemical Industries. According to the manufacturer, the aqueous silica colloid possessed a silica content of 30% by wt., pH of 3, and silica particle size of 12 nanometers.All organosilanes were used as received from the manufacturer. Methylacryloxypropyltrimethoxysilane (MPTMS) and acryl-oxypropyltrimethoxysilane (APTMS) were obtained from Aldrich Chemical and United Chemical Technologies, respectively, and acetoxypropyltrimethoxysilane (AcPTMS) and methoxypropyl-trimethoxysilane (MePTMS) were obtained from Gelest Inc. Isopropanol (IPA), 1-methoxy-2-propanol (PM), and propylene glycol methyl ether acetate (PMA) were used as received from Aldrich Chemical.
The procedure for producing the ORMOSIL sols was as follows: 1,500 g of Snowtex ST-O-30 was added to rapidly stirring IPA. 3.15 g of a 33% by wt. solution of a proprietary free radical scavenger in PM was added to the colloidal silica/IPA mixture. The mixture was then split into 10 240-g aliquots. To each aliquot, an organosilane was quickly added with rapid stirring. The chemical composition and level of organosilane added to each aliquot is shown in Table 1. Each sol was refluxed for 2 hours, cooled to room temperature, and then 100 g of PM and 100 g of PMA were added. Next, each sol was concentrated by about 50% by vacuum stripping solvent using a rotary evaporator. As determined by proton nuclear magnetic resonance spectroscopy, the ORMOSIL sols were suspended primarily in PMA since PMA was the highest boiling solvent in the sols. The solids content of the resulting ORMOSIL colloids was determined gravimetrically by evaporating all volatiles using a vacuum oven. The solids content of each colloid is shown in Table 1.
Three different oligomeric acrylates were used as polymer matrix precursors for the ORMOSILs of interest. EB220, an aromatic, hexafunctional urethane acrylate (UHA), and RDX51027, a brominated epoxy diacrylate (EDA), were used as received from UCB Chemical. An aliphatic urethane diacrylate (UDA) produced from 3-isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate (IPDI) and polycaprolactone diol was synthesized as follows.
To a 3-neck flask equipped with an addition funnel, temperature probe, and mechanical stirrer was charged 33.35 g of IPDI (Bayer Corp.) and 50 mg dibutyltin dilaurate (Aldrich Chemical). The addition funnel was charged with 39.75 g of warm (~ 40 deg C) polycaprolactone diol (530 g/mol from Aldrich Chemical), which was added to the contents of the flask at 55-60 deg C. The mixture was stirred at 65 deg C for 14 hrs. The temperature was then cooled to 55 deg C before a mixture of 20.07 g of hydroxyethyl acrylate (Aldrich Chemical) and 100 mg of hydroquinone monomethyl ether was added while maintaining the temperature between 54 and 58 deg C. This mixture was stirred at 55 deg C for 10-12 hrs while completion of the reaction was verified by infrared spectroscopy. The resulting colorless, viscous liquid was used without further purification.
Coating Preparation, Application and Curing
Coatings were prepared by dissolving the oligomeric acrylate in PM, adding the appropriate amount of ORMOSIL sol, and then adding the appropriate amount of a 10% by wt. solution of photoinitiator (Darocur 4265 from Ciba Chemicals) in PM. The amount of photoinitiator used was 0.5 wt. % based on the total weight of coating solids. The coating solutions were designed to be about 40% solids with 0.5 wt. % photoinitiator based on total solids.Samples for measuring optical clarity, abrasion resistance, and adhesion were prepared by spin coating the coating solutions onto 4x4x1/8-in. pieces of polycarbonate sheet (Lexan[R] 9030 from GE Plastics). The coating process consisted of cleaning the surface of the polycarbonate with isopropanol by squirting isopropanol onto the polycarbonate surface while spinning, allowing the isopropanol to evaporate, dispensing approximately 1 mL of coating solution to the center of the plaque, and then spinning at a speed of 3,000 rpm for 20 sec. The spin-coated samples were immediately cured in a nitrogen atmosphere using a Fusion model DRS-120 ultraviolet processor. A 600-watt H bulb was used in conjunction with a belt speed of 6.5 cm/sec and lamp to sample distance of 2 in. to cure the coatings. Each coating sample was passed three times through the processor. A high degree of cure was confirmed by observing the disappearance of the vibrational absorbances associated with the vinyl groups of the uncured coating using infrared spectroscopy.
Free films for dynamic mechanical analysis (DMA) were prepared by pouring approximately two grams of coating solution into an aluminum pan 5 cm in diameter and placing the pan into a forced air oven at 70 deg C to evaporate the solvent. The weight of the sample was monitored to ensure essentially complete solvent removal. The solventless material was then UV cured in the pan under a nitrogen blanket. Specimens for DMA were cut from the cured circular films.
Characterization
The optical clarity of the coatings was determined by measuring the haze of the coated polycarbonate samples according to ASTM D 1003. Abrasion resistance and adhesion was determined on the coated polycarbonate samples using the Taber Abrasion Test (ASTM D 1044) and Tape Test (ASTM 3359), respectively. DMA was conducted on free films about 0.25 mm thick using a Rheometrics Solids Analyzer RSAII equipped with a rectangular tension fixture. A frequency of 1 rad/sec, strain of 0.01%, and temperature ramp of 2 deg C/min. was used.Proton nuclear magnetic resonance spectroscopy (1H NMR) was conducted using a Bruker Avance 400 NMR spectrometer operating at 400.132 MHz using a 5 mm carbon-proton dual probe. A sweep width of 8300 Hz was used with 32k data points, resulting in an acquisition time of 3.958 sec. A pulse delay of 1 sec was also used, as was a flip angle of 30 deg . Sixteen scans were obtained using two dummy scans. Processing parameters included apodization with 0.30 Hz line broadening, automatic phase adjustment, and baseline correction.
Transmission electron microscopy (TEM) was conduced with the use of a Philips CM100 microscope operated at 100 kV. Materials were prepared by ultramicrotomy performed at room temperature.
Results and Discussion
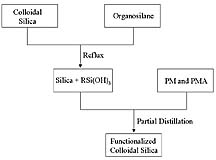
The process used for preparing the ORMOSILs is shown schematically in Figure 1. The aqueous, acidic colloidal silica was diluted with IPA and the organosilane was quickly added to the rapidly stirring colloid at room temperature. The solution was then refluxed for two hours. During this step, the organosilane was hydrolyzed to form silanol groups and some condensation of silanols may have occurred.9 Next, the mixture of colloidal silica and hydrolyzed silane was diluted with a 50/50 PM/PMA solvent mixture. Dilution with this solvent mixture enabled the next step of the process, which involved increasing the degree of silanol condensation by removal of water and IPA from the system. PM is higher boiling than both water and IPA and forms a 50% azeotrope with water, which promotes condensation by removal of water from the system and increasing the boiling temperature.10 PMA was added to the system to further promote the level of silanol condensation. PMA is miscible with PM and IPA and higher boiling than both PM and IPA. Water has very limited solubility in PMA (3 wt. %). Thus, PMA allows for a higher temperature to be reached during vacuum stripping and promotes the water removal required to drive silanol condensation. With this process, vacuum distillation results in complete removal of water, IPA, and PM to produce the functionalized silica colloid in PMA. H1 NMR was used to confirm the composition of the solvent system of the colloid.Since the functional groups present in the organosilane are susceptible to polymerization and/or hydrolysis or alcoholysis, it was important to determine the fate of these functional groups resulting from the ORMOSIL synthesis process. 1H NMR was used to observe any processing induced chemical modifications of the organic group of the ORMOSILs. Specifically, the methylene protons of the n-propyl group were used to determine if the functional group underwent hydrolysis or alcoholysis during the functionalization process.
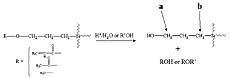
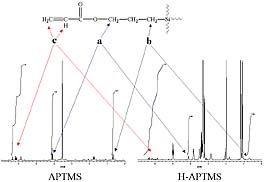
Hydrolysis or alcoholysis would result in the production of a hydroxyl group next to the methylene group (“a”) as shown in Figure 2. This change in structure produced by hydrolysis or alcoholysis would result in a significant chemical shift for these methylene protons (“a”). For example, the chemical shift for the methylene protons next to the oxygen atom in n-propanol is 0.26 PPM down field and 0.43 PPM up field from the analogous methylene protons of methyl propyl ether and propyl acetate, respectively.11 In contrast, hydrolysis or alcoholysis of the functional group would not result in a significant chemical shift for the methylene protons adjacent to the silicon atom (“b” in Figure 2) since these methylene protons are four atoms removed from the functional group. Thus, integration of methylene protons “a” and “b” allowed for the determination of the extent of hydrolysis or alcoholysis produced during ORMOSIL preparation. Figure 3 displays the 1H NMR spectra for APTMS and the H-APTMS ORMOSIL illustrating the peaks used for the integration.
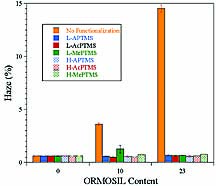
Table 2 displays the chemical shift and integration data obtained from the analysis for hydrolysis or alcoholysis of the organic group. The data does not show any evidence of hydrolysis or alcoholysis for either APTMS or AcPTMS-based ORMOSILs.
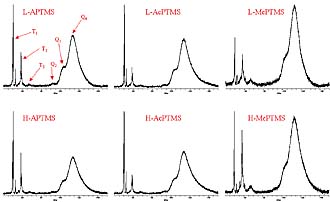
In addition to determining the fate of the organic functional groups, it was important to determine the extent of silanol condensation produced during the functionalization process as well as the nature of the condensed species. 29Si NMR was used to obtain the information desired. For species derived from trifunctional organosilanes, four groups of peak Si absorptions can be observed in the 29Si NMR spectra. Peaks associated with these trifunctional silicon species are usually referred to as T peaks. T0, T1, T2, and T3 correspond to trifunctional silicon species that have undergone 0, 1, 2, and 3 condensation reactions, respectively (see Figure 4). The typical chemical shift ranges for these T peaks is: T0: Ñ37 to Ð39 ppm, T1: Ñ46 to Ð 48 ppm, T2: Ñ53 to Ð57 ppm, and T3: Ñ61 to Ð66 ppm.12-14
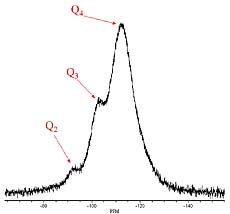
Figure 5 displays the 29Si NMR spectra for all of the ORMOSILs produced. The lack of a group of peaks at Ð40 ppm indicates that at least one silanol per organosilane molecule underwent a condensation reaction. The minor group of peaks just upfield from the intense group of peaks at Ð50 ppm was believed to be due to monocondensed species containing an alkoxy group obtained by condensation with protic solvent molecules, IPA or PM.15 Table 3 lists integration results that describe the distribution of condensed species in the ORMOSILs. The results show that the distribution of T species was similar for APTMS and AcPTMS-based ORMOSILs while the use of MePTMS resulted in a much higher level of T2 and T3 species.

Insight into the degree of organosilane-silica condensation was obtained by considering the broad group of peaks for the 29Si absorption associated with colloidal silica. These 29Si peaks are typically referred to as the “Q” peaks and can be split into five different groups of peaks (Q0, Q1, Q2, Q3, Q4), which differ by the degree of condensation around the silicon atom. A Q0 peak indicates no condensation of OH or OR groups around the silicon atom while Q1- Q4 indicate one to four condensed silanols around the silicon atom as shown in Figure 4. Typical Q chemical shift ranges are: Q0: Ñ72 to Ð82 ppm, Q1: Ñ82 to Ð 89 ppm, Q2: Ñ92 to Ð96 ppm, Q3: Ñ100 to Ð104 ppm, and Q4: ~Ñ110 ppm.
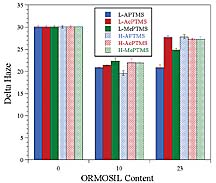
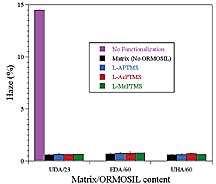
For clear coatings, optical clarity of the coating is very important. Typically, the optical clarity of a coating on a transparent substrate is determined with a hazemeter according to ASTM D 1003. The hazemeter measures the ratio of diffuse transmitted light to total transmitted light. The affect of organic functionalization of the silica nanoparticles on optical clarity was determined by preparing control coatings based on unfunctionalized silica. The unfunctionalized silica sol was processed the same as that of the ORMOSIL sols except that no organosilane was added. As shown in Figure 7, organic functionalization was found to be crucial for producing coatings with high optical clarity. With regard to the chemical composition and level of organosilane functionalization, with the exception of coatings derived from the MePTMS sols, all of the coatings showed essentially the same haze as the matrix polymer (UDA). The slightly higher haze for coatings derived from the MePTMS sols suggests the presence of some slight silica agglomeration. The higher haze level observed for the MePTMS-derived UDA coatings may have been the result of the lower level of silica functionalization observed for the MePTMS-based ORMOSILs (see Table 4). Interestingly, for the other matrix acrylates, the MePTMS ORMOSILs gave essentially the same excellent optical clarity as the other ORMOSILs.
The very high optical clarity of the ORMOSIL-containing coatings indicates excellent dispersion of the ORMOSIL nanoparticles. Nanoparticle dispersion was characterized using transmission electron microscopy (TEM). The coatings used for the morphological characterization were based on UHA and 60 wt. % ORMOSIL functionalized at the lower level employed. As expected based on the high optical clarity, nanoparticle dispersion was very homogenous and independent of the nature of the organosilane utilized. Figure 9 displays a representative TEM micrograph obtained.
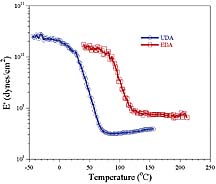
A wide array of test methods have been used for evaluating the abrasion resistance of coatings. The industry standard method for evaluating clear coatings on transparent, plastic substrates is the Taber abrasion test (TAT). With this method, a pair of abrasive-imbedded wheels track around the coated surface as the sample rotates for a specified number of revolutions. The degree of damage imparted to the surface is represented by the change in haze induced by the abrasion process. It is known that the viscoelastic properties and adhesion of a coating strongly affect the results of the TAT, but a complete understanding of the coating composition-Taber abrasion relationship is far from being understood.16 As discussed previously, the matrix forming, oligomeric acrylates were chosen to provide a range of viscoelastic properties from lightly crosslinked, rubbery materials to highly crosslinked, glassy materials. In order to more precisely describe the viscoelastic properties of the matrix materials, free films were prepared and characterized by DMA. Unfortunately, the film produced from UHA was too brittle to be measured with DMA. Figure 10 displays the storage moduli versus temperature behavior of cured films of UDA and EDA. The results show that at ambient temperature UDA is much lower modulus than EDA since the Tg of UDA is close to ambient temperature while EDA is clearly in the glassy state at this temperature.

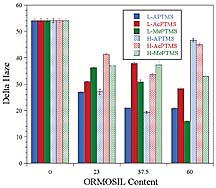
For the rubbery UDA matrix, reinforcement with ORMOSIL nanoparticles resulted in an improvement in abrasion resistance and the magnitude of the improvement was dependent on the level of ORMOSIL. For this matrix, the abrasion resistance was found to decrease as the ORMOSIL concentration was increased from 10 to 23 wt. % indicating the existence of an optimum ORMOSIL concentration below 23 wt. %. The high optical clarity observed for the 23 wt. % ORMOSIL modified UDA coatings suggested a homogenous dispersion of nanoparticles and, thus, good compatibility between the matrix and the ORMOSIL nanoparticles. As a result, the decrease in abrasion resistance observed with an increase in ORMOSIL content to 23 wt. % was somewhat curious since the hardness and, thus, the abrasion resistance should have increased. As mentioned, abrasion resistance as determined by the TAT is influenced not only by the viscoelastic properties of the coating but also by the adhesive properties. Chisholm et al.17 investigated the effect of adhesion on the Taber abrasion results by preparing silicone coatings and coating them on polycarbonate with and without a primer layer. The results showed an effect of the primer with the abrasion resistance being markedly higher for the primed samples. Using florescence microscopy, it was shown that the mechanism of abrasion from the primed samples consisted of scratching and marring of the coating (cohesive failure) while the mechanism of abrasion for the unprimed samples also included chipping or delamination of the coating from the substrate due to adhesion failure.
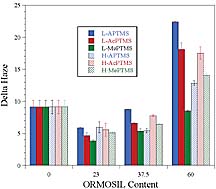
In addition, to crosshatch adhesion, the adhesion of the UDA-based coating was evaluated by a simple tape pull test (no crosshatch pattern). At ORMOSIL levels of 0 and 10 wt. %, no coating was removed by this method, but, at 23 wt. % ORMOSIL, two coatings failed. This result is somewhat consistent with the hypothesis that the reduction in abrasion resistance observed as the ORMOSIL content was increased from 10 to 23 wt. % was due to a reduction in the degree of adhesion between the substrate and coating. Similar to the UDA-based coatings, UHA-based coatings exhibited an optimum in abrasion resistance with ORMOSIL content. For this matrix, the highest abrasion resistance was observed at 23 wt. % ORMOSIL. Overall, the UHA coatings showed the highest abrasion resistance, which was attributed to the excellent adhesion obtained with these coatings and the high crosslink density resulting from the high acrylate content of the UHA.
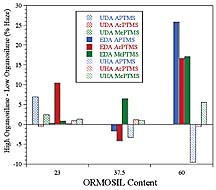
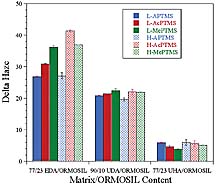
ORMOSILs with acrylate groups would result in the production of a higher crosslink density as compared to analogous coatings produced from nonreactive ORMOSILs. Higher crosslink density should translate to higher hardness and modulus, which, assuming adequate adhesion to the substrate, should result in an increase in abrasion resistance. However, if the crosslink density is extremely high, the coating may become very brittle resulting in delamination of the coating from the substrate due to the reduced ability of the coating to dissipate the stresses imparted by the abrasion process. A brittle coating may result in greater transfer of stress to the coating-substrate interface enhancing the chance for adhesive failure. A comparison of the abrasion resistance results obtained for a given matrix acrylate, ORMOSIL content, and organosilane indicated that increasing the level of organosilane used in the functionalization process by 50% from 1.04 x 10-3 mol per gram silica to 1.56 x 10-3 mol per gram silica, in general, did not improve abrasion resistance. In fact, as illustrated in Figure 16, increasing the level of organosilane produced a trend, albeit a weak one, toward a reduction in abrasion resistance. Since the coatings were compared at equivalent ORMOSIL content, this trend could be due to the fact that the silica level of the ORMOSILs produced with the high level of organosilane functionalization was 5% lower than those produced with the low level of organosilane. Clearly, from an abrasion resistance standpoint, it is desirable to limit the amount of organosilane functionalization to a level just sufficient to allow for homogeneous dispersion of the nanoparticles in the matrix of interest.
Conclusion
The results of this study showed that the addition of organically modified silica particles to oligomeric acrylates can result in the production of UV curable coatings that possess high optical clarity, excellent adhesion to polycarbonate, and improved abrasion resistance. However, the magnitude of the property enhancement provided by the ORMOSIL was very dependent on several variables such as the organic composition of the ORMOSIL, the level of ORMOSIL, and the chemical composition of the polymer matrix. With regard to organic modification of silica, it was found that organic modification was essential for obtaining coatings with high optical clarity. For the coatings derived from the difunctional acrylate oligomers, UDA and EDA, the acrylate functional ORMOSILs provided the highest abrasion resistance while the opposite trend was observed for the hexafunctional acrylate oligomer, UHA. This behavior was attributed to an optimum balance between coating hardness and adhesion. In general, increasing coating hardness or modulus should result in an increase in abrasion resistance. However, if the hardness or modulus is extremely high, the abrasion process may result in delamination of the coating from the substrate due to greater transfer of stress to the coating-substrate interface. For the three matrix-forming oligomers investigated, the relative degree of hardness for the cured matrices was EHA>EDA>UDA as suggested from DMA and abrasion resistance results. For the more lightly crosslinked matrix oligomers, UDA and EDA, the acrylate functional ORMOSILs provided the highest abrasion resistance because of the higher crosslink density achieved by grafting the silica particles to the organic matrix. In contrast, for the more highly crosslinked matrix derived from EHA, grafting of the silica nanoparticles to the brittle matrix may have caused a greater degree of adhesive failure during the abrasion process and thus reduced abrasion resistance.The rationale used to explain the abrasion resistance results obtained as a function of ORMOSIL chemical composition can also be used to explain the results obtained as a function of ORMOSIL content. In general, the abrasion resistance data obtained as a function of ORMOSIL content showed the existence of an optimum ORMOSIL content that was dependent on the matrix composition. Displacing organic matrix material with silica increases coating hardness but decreases adhesion to the polymer substrate. Thus, an optimum ORMOSIL content for a given matrix would be the highest level that does not result in delamination of the coating during the abrasion process.
Acknowledgement
The authors would like to thank Eric Olson, Thomas Early, and Greg Goddard for conducting electron microscopy, 29Si NMR, and IR spectroscopy, respectively, and Elizabeth Williams and Peter Codella for helpful discussions.This article is based on a paper presented at the International Waterborne, High-Solids, and Powder Coatings Symposium February 26-28, 2003, in New Orleans, sponsored by The University of Southern Mississippi Department of Polymer Science.
For more information, contact Bret Chisholm, GE Corporate Research and Development, One Research Circle, Niskayuna, NY 12309.
References
1 Wright, A.P. US Patent 5,374,483.2 Philipp, G.; Schmidt, H. J. Non-Cryst. Solids, 63, 283(1984).
3 Wilkes, G.L.; Orler, B.; Huang, H. Polym. Prepr., 26, 300(1985).
4 Young, S.K.; Jarrett, W.L.; Mauritz, K.A.Polymer, 43, 2311 (2002).
5 Metroke, T.L.; Kachurina, O.; Knobbe, E.T. Progress in Organic Coatings, 44(4), 295 (2002).
6 Chu, L.; Daniels, M.W.; Francis, L.F. Mat. Res. Soc. Symp. Proc., 435, 221 (1996).
7 Hunnicutt, M.L.; Harris, J.M. Anal. Chem., 58, 748 (1986).
8 Becker, C.; Krug, H.; Schmidt, H. Mat. Res. Soc. Symp. Proc., 435, 237 (1996).
9 Brinker, C.J.; Scherer, G.W. Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, Academic Press, San Diego, CA, 1990.
10 Flick, E.W. Industrial Solvents Handbook 4th Edition, Noyes Data Corp., Park Ridge, NJ, 1991, p. 576.
11 S. Sasaki, Handbook of Proton-NMR Spectra and Data Volume 1, Academic Press, New York., 1985.
12 F. Babonneau, K. Thorne, J. D. Mackenzie, Chem. Mater., 1(5), 554 (1989).
13 Peeters, M.P.J.; Wakelkamp, W.J.J.; Kentgens, A.P.M. J. Non-Cryst Solids, 189, 11 (1995).
14 Alam, T.M.; Assink, R.A.; Loy, D.A. Chem. Mater., 8, 2366 (1996).
15 Nishiyama, N.; Asakura, T.; Horie, K. J. Colloid Int. Sci., 124, 14 (1988).
16 Sun, J.; Mukamal, H.; Liu, Z.; Shen, W. Tribology Letter, 13, 49 (2002).
17 Chisholm, B. J.; Potyrailo, R.; Shaffer, R.; Cawse, J.; Brennan, M.; Molaison, C. Proc. Int. Coatings for Plastis Symp., 5, (2002).
Looking for a reprint of this article?
From high-res PDFs to custom plaques, order your copy today!





